Run time of analysis
Givanna Putri
2023-06-13
Last updated: 2023-08-15
Checks: 7 0
Knit directory: SuperCellCyto-analysis/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(42) was run prior to running the
code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 7baf7b7. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Ignored: code/b_cell_identification/.DS_Store
Ignored: code/batch_correction/.DS_Store
Ignored: code/explore_supercell_purity_clustering/.DS_Store
Ignored: code/explore_supercell_purity_clustering/functions/.DS_Store
Ignored: code/explore_supercell_purity_clustering/louvain_all_cells/.DS_Store
Ignored: code/label_transfer/.Rhistory
Ignored: data/.DS_Store
Ignored: data/bodenmiller_cytof/
Ignored: data/explore_supercell_purity_clustering/
Ignored: data/haas_bm/
Ignored: data/oetjen_bm_dataset/
Ignored: data/trussart_cytofruv/
Ignored: output/.DS_Store
Ignored: output/bodenmiller_cytof/
Ignored: output/explore_supercell_purity_clustering/
Ignored: output/label_transfer/
Ignored: output/oetjen_b_cell_panel/
Ignored: output/trussart_cytofruv/
Untracked files:
Untracked: code/b_cell_identification/cluster_singlecell.R
Untracked: code/b_cell_identification/rescale_supercell.R
Untracked: code/b_cell_identification/runtime_benchmark/
Untracked: code/batch_correction/benchmark_cycombine.R
Untracked: code/batch_correction/rescale_supercell.R
Untracked: code/batch_correction/runtime_benchmark/
Untracked: code/bodenmiller_data/benchmark_supercell_runtime.R
Untracked: code/compare_da_test_runtime.R
Untracked: code/krieg_melanoma/
Untracked: code/label_transfer/harmony_knn_singlecell.R
Untracked: code/label_transfer/seurat_rpca_singlecell.R
Untracked: output/krieg_melanoma/
Unstaged changes:
Modified: SuperCellCyto-analysis.Rproj
Modified: code/batch_correction/run_batch_correction_supercells.R
Modified: code/label_transfer/harmony_knn.R
Modified: code/label_transfer/seurat_rpca.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/run_time.Rmd) and HTML
(docs/run_time.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 7baf7b7 | Givanna Putri | 2023-08-15 | wflow_publish("analysis/*") |
| html | a55c3ba | Givanna Putri | 2023-07-28 | Build site. |
| html | 366514e | Givanna Putri | 2023-07-28 | Build site. |
| Rmd | 402358b | Givanna Putri | 2023-07-28 | wflow_publish(c("analysis/*Rmd")) |
Introduction
In this analysis, we examine the time required to create the supercells, as well as the run time improvement obtained by analysing supercells vs single cells.
library(here)
library(data.table)
library(stringr)
library(ggplot2)
library(scales)
library(viridis)Function to convert the log 10 y-axis scale to actual number. Just so it’s easier to interpret. So instead of showing 1,2,3, which are the log 10 values, we show 10^1, 10^2, 10^3, etc.
convert_log10_to_actual <- function(labels, dp_below_1 = 2) {
transformed_labs <- sapply(labels, function(x) {
new_lab <- 10^x
# if over 1, then round to integer, otherwise round to whatever dp_below_1 is
if (new_lab >= 1) {
new_lab <- round(new_lab)
} else {
new_lab <- round(new_lab, dp_below_1)
new_lab <- sprintf(paste0("%.", dp_below_1, "f"), new_lab)
}
return(new_lab)
})
}Supercell creation
datasets <- c("samusik_all", "levine_32dim")
supercell_runtime <- lapply(datasets, function(dt_source) {
dt <- fread(here("output", "explore_supercell_purity_clustering", "20230511",
dt_source, "supercell_runs", "supercell_runtime.txt"),
sep = ":", header = FALSE, col.names = c("dataset", "duration_seconds"))
dt$duration_seconds <- gsub(" sec elapsed", "", dt$duration_seconds)
mean_duration <- mean(as.numeric(dt$duration_seconds))
return(mean_duration)
})
names(supercell_runtime) <- str_to_title(datasets)run_time_bcells <- fread(here("output", "oetjen_b_cell_panel", "20230511", "supercell_runtime.txt"),
sep = ":", header = FALSE, col.names = c("dataset", "duration_seconds"))
supercell_runtime[["Oetjen_bcells"]] <- mean(as.numeric(gsub(" sec elapsed", "", run_time_bcells$duration_seconds)))run_time_trussart <- fread(here("output", "trussart_cytofruv", "20230515_supercell_out", "supercell_runtime.txt"),
sep = ":", header = FALSE, col.names = c("dataset", "duration_seconds"))
supercell_runtime[["Trussart_cytofruv"]] <- mean(as.numeric(gsub(" sec elapsed", "", run_time_trussart$duration_seconds)))run_time_bodenmiller <- fread(here("output", "bodenmiller_cytof", "supercell_benchmark.txt"),
sep = ":", header = FALSE, col.names = c("dataset", "duration_seconds"))
supercell_runtime[["BCR_XL"]] <- mean(as.numeric(gsub(" sec elapsed", "", run_time_bodenmiller$duration_seconds)))run_time_krieg <- fread(here("output", "krieg_melanoma", "supercell_benchmark.txt"),
sep = ":", header = FALSE, col.names = c("dataset", "duration_seconds"))
supercell_runtime[["Anti_PD1"]] <- mean(as.numeric(gsub(" sec elapsed", "", run_time_krieg$duration_seconds)))supercell_runtime_dt <- data.table(
dataset=names(supercell_runtime),
duration_seconds=as.numeric(supercell_runtime)
)
supercell_runtime_dt[, duration_minutes := duration_seconds/60]
supercell_runtime_dt[, log10_duration_minutes := log10(duration_minutes)]
# manually entered this so watch out if it is wrong
supercell_runtime_dt[, ncells := c(841644, 265627, 8314260, 8589739, 172791, 85715)]
# mainly for plotting so we plot from smallest to largest
supercell_runtime_dt <- supercell_runtime_dt[order(ncells)]
supercell_runtime_dt[, ncells_pretty_label := prettyNum(ncells, big.mark=",", scientific=FALSE)]
supercell_runtime_dt[, ncells_pretty_label := factor(ncells_pretty_label, levels=supercell_runtime_dt$ncells_pretty_label)]y_axis_labs <- seq(-1.5,2.5,0.5)
ggplot(supercell_runtime_dt, aes(x = ncells_pretty_label, y = log10_duration_minutes)) +
geom_point(aes(colour = dataset), size = 2) +
theme_classic() +
scale_y_continuous(breaks = y_axis_labs, label = convert_log10_to_actual(y_axis_labs,1)) +
labs(y = "Duration (minutes)", x = "Number of cells", color = "Dataset", title = "Time taken to create supercells") +
theme(axis.text.x = element_text(angle = 45, hjust=1), legend.position="bottom")
| Version | Author | Date |
|---|---|---|
| 366514e | Givanna Putri | 2023-07-28 |
Batch correction
cycombine_duration_run1 <- fread("output/trussart_cytofruv/benchmark_duration/cycombine_benchmark_run1.txt")
cycombine_duration_run1[, run := 'run_1']
cycombine_duration_run2 <- fread("output/trussart_cytofruv/benchmark_duration/cycombine_benchmark_run2.txt")
cycombine_duration_run2[, run := 'run_2']
cycombine_duration <- rbind(cycombine_duration_run1, cycombine_duration_run2)
cycombine_duration <- cycombine_duration[, c("V6","V4", "V7", "run")]
names(cycombine_duration) <- c("gamma", "setting", "duration_seconds", "run")
cycombine_duration[, gamma := gsub(":", "", gamma)]
cycombine_duration[, gamma := gsub("singlecell", "single cell", gamma)]
cycombine_duration[, gamma := gsub("gam", "", gamma)]
cycombine_duration <- cycombine_duration[, .(duration_seconds = mean(duration_seconds)), by=c('gamma', 'setting')]
cycombine_duration[, algorithm := 'cyCombine']cytofruv_duration_singlecell <- lapply(seq(24), function(setting) {
dt_run1 <- fread(paste0("output/trussart_cytofruv/benchmark_duration/cytofruv/run1/cytofruv_singlecell_setting", setting, ".txt"))
dt_run2 <- fread(paste0("output/trussart_cytofruv/benchmark_duration/cytofruv/run2/cytofruv_singlecell_setting", setting, ".txt"))
mean_duration <- mean(c(as.numeric(dt_run1$V5), as.numeric(dt_run2$V5)))
return(mean_duration)
})
cytofruv_duration_singlecell_dt <- data.table(
gamma = "single cell",
setting = seq(24),
duration_seconds = cytofruv_duration_singlecell
)
cytofruv_duration_supercell <- lapply(seq(10,40,10), function(gamma_val) {
cytofruv_duration <- lapply(seq(24), function(setting) {
dt_run1 <- fread(paste0("output/trussart_cytofruv/benchmark_duration/cytofruv/run1/cytofruv_supercell_gam", gamma_val, "_setting", setting, ".txt"))
dt_run2 <- fread(paste0("output/trussart_cytofruv/benchmark_duration/cytofruv/run2/cytofruv_supercell_gam", gamma_val, "_setting", setting, ".txt"))
mean_duration <- mean(c(as.numeric(dt_run1$V8), as.numeric(dt_run2$V8)))
return(mean_duration)
})
cytofruv_duration_dt <- data.table(
gamma = gamma_val,
setting = seq(24),
duration_seconds = cytofruv_duration
)
return(cytofruv_duration_dt)
})
cytofruv_duration_supercell[['singlecell']] <- cytofruv_duration_singlecell_dtbatch_correction_duration <- rbindlist(cytofruv_duration_supercell)
batch_correction_duration[, algorithm := "CytofRUV"]
batch_correction_duration <- rbind(batch_correction_duration, cycombine_duration)
batch_correction_duration[, algorithm := factor(algorithm)]
batch_correction_duration[, gamma := factor(gamma, levels = c("single cell", seq(5,50,5)))]
batch_correction_duration[, duration_seconds := as.numeric(duration_seconds)]
batch_correction_duration[, duration_minutes := duration_seconds/60]
batch_correction_duration[, log10_duration_minutes := log10(duration_minutes)]y_axis_labs <- seq(-1.2,1.2,0.2)
ggplot(batch_correction_duration, aes(x = gamma, y = log10_duration_minutes, fill=algorithm)) +
geom_boxplot(outlier.size=0.2, lwd=0.1) +
scale_fill_brewer(palette="Dark2") +
theme_classic() +
facet_wrap(~algorithm) +
theme(axis.text.x = element_text(angle = 45, hjust=1), legend.position="none") +
scale_y_continuous(breaks = y_axis_labs, label = convert_log10_to_actual(y_axis_labs)) +
labs(y = "Duration (minutes)", x = "Gamma", title = "Time taken to do batch correction", fill='Algorithm')
Louvain clustering
louvain_runtime_supercell <- lapply(c("samusik_all", "levine_32dim"), function(dt_source) {
dt <- lapply(seq(5, 50, by=5), function(gam) {
dt <- fread(here("output", "explore_supercell_purity_clustering", "20230511",
dt_source, "louvain_supercell_runs", paste0("gamma_", gam),
"louvain_supercell_runtime.txt"))
res <- data.table(
setting = factor(sapply(dt$V1, function(x) str_split_1(x, "_")[3])),
duration_seconds = as.numeric(dt$V2)
)
med_dur <- res[, .(med_dur = mean(duration_seconds)), by = c("setting")]
res <- data.table(
gamma = gam,
setting = med_dur$setting,
duration_seconds = as.numeric(med_dur$med_dur),
dataset = dt_source
)
return(res)
})
return(rbindlist(dt))
})
louvain_runtime_supercell <- rbindlist(louvain_runtime_supercell)
louvain_runtime_allcell <- lapply(c("samusik_all", "levine_32dim"), function(dt_source) {
dt <- lapply(seq(10, 30, by=5), function(k) {
dt <- fread(here("output", "explore_supercell_purity_clustering", "20230511",
dt_source, "louvain_allcells", paste0("k", k),
"louvain_supercell_runtime.txt"))
res <- data.table(
gamma = "single cell",
setting = sapply(dt$V1, function(x) str_split_1(x, "_")[3])[1],
duration_seconds = mean(as.numeric(dt$V2)),
dataset = dt_source
)
return(res)
})
return(rbindlist(dt))
})
louvain_runtime_allcell <- rbindlist(louvain_runtime_allcell)
louvain_runtime_others <- rbind(
louvain_runtime_allcell,
louvain_runtime_supercell
)k_vals <- c(3,5,10,15,20)
oetjen_duration_singlecell <- lapply(k_vals, function(setting) {
dt <- fread(paste0("output/oetjen_b_cell_panel/benchmark_runtime/run1/duration_louvain_singlecell_k", setting, ".txt"))
return(as.numeric(dt$V2))
})
oetjen_duration_singlecell_dt <- data.table(
gamma = "single cell",
setting = k_vals,
duration_seconds = oetjen_duration_singlecell
)
oetjen_duration_supercell <- lapply(seq(10,40,10), function(gamma_val) {
oetjen_duration <- lapply(k_vals, function(setting) {
dt <- fread(paste0("output/oetjen_b_cell_panel/benchmark_runtime/run1/duration_louvain_supercell_gamma", gamma_val, "_k", setting, ".txt"))
return(as.numeric(dt$V2))
})
oetjen_duration_dt <- data.table(
gamma = gamma_val,
setting = k_vals,
duration_seconds = oetjen_duration
)
return(oetjen_duration_dt)
})
oetjen_duration_supercell[['singlecell']] <- oetjen_duration_singlecell_dtoetjen_duration_dt <- rbindlist(oetjen_duration_supercell)
oetjen_duration_dt[, dataset := "oetjen_bcells"]
louvain_runtime <- rbind(louvain_runtime_others, oetjen_duration_dt)
louvain_runtime[, duration_seconds := as.numeric(duration_seconds)]
louvain_runtime[, log10_duration_seconds := log10(duration_seconds)]
louvain_runtime[, duration_minutes := duration_seconds/60]
louvain_runtime[, log10_duration_minutes := log10(duration_minutes)]
louvain_runtime[, gamma := factor(gamma, levels = c("single cell", seq(5,50,5)))]y_axis_labs <- seq(-2,3.5,0.5)
ggplot(louvain_runtime, aes(x = gamma, y = log10_duration_minutes, fill=dataset)) +
geom_boxplot(outlier.size = 0.1, lwd=0.1) +
scale_fill_viridis(discrete = TRUE, alpha=0.6) +
theme_classic() +
facet_wrap(~dataset) +
scale_y_continuous(breaks = y_axis_labs, label = convert_log10_to_actual(y_axis_labs)) +
labs(y = "Duration (minutes)", x = "Gamma", title = "Time taken to run Louvain clustering", fill="Dataset") +
theme(axis.text.x = element_text(angle = 45, hjust=1), legend.position="none")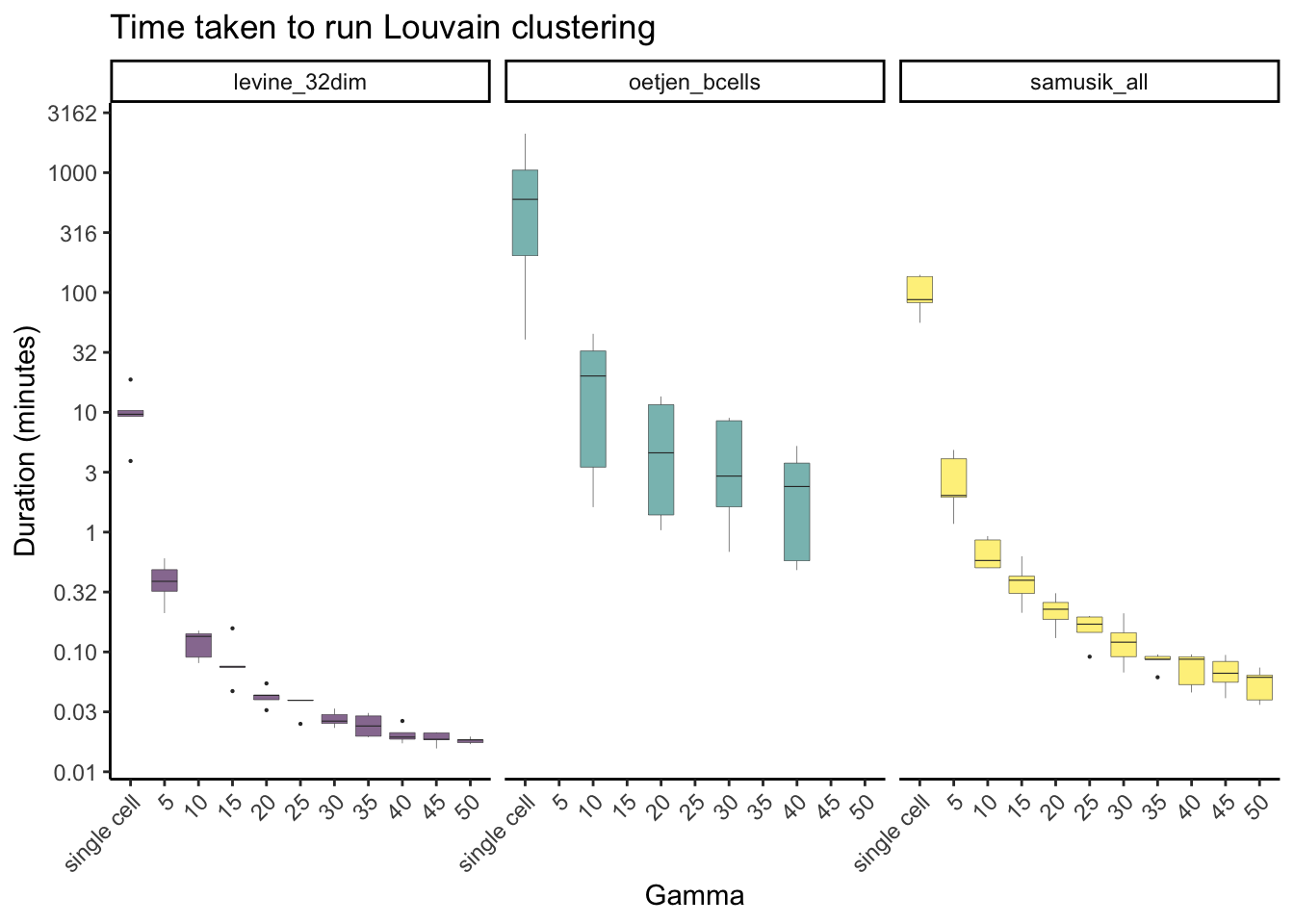
FlowSOM clustering
fsom_runtime_supercell <- lapply(c("samusik_all", "levine_32dim"), function(dt_source) {
dt <- lapply(seq(5, 50, by=5), function(gam) {
dt <- fread(here("output", "explore_supercell_purity_clustering", "20230511",
dt_source, "flowsom_supercell_runs", paste0("gamma_", gam),
"flowsom_supercell_runtime.txt"))
res <- data.table(
setting = factor(sapply(dt$V1, function(x) paste0(str_split_1(x, "_")[3], "_", str_split_1(x, "_")[4]))),
duration_seconds = as.numeric(dt$V2)
)
med_dur <- res[, .(med_dur = mean(duration_seconds)), by = c("setting")]
res <- data.table(
gamma = gam,
setting = med_dur$setting,
duration_seconds = as.numeric(med_dur$med_dur),
dataset = dt_source
)
return(res)
})
return(rbindlist(dt))
})
fsom_runtime_supercell <- rbindlist(fsom_runtime_supercell)
fsom_runtime_allcell <- lapply(c("samusik_all", "levine_32dim"), function(dt_source) {
dt <- fread(here("output", "explore_supercell_purity_clustering", "20230509",
dt_source, "flowsom_allcells", "flowsom_allcell_runtime.txt"))
res <- data.table(
setting = factor(sapply(dt$V1, function(x) paste0(str_split_1(x, "_")[3], "_", str_split_1(x, "_")[4]))),
duration_seconds = as.numeric(dt$V2)
)
med_dur <- res[, .(med_dur = mean(duration_seconds)), by = c("setting")]
res <- data.table(
gamma = "single cell",
setting = med_dur$setting,
duration_seconds = as.numeric(med_dur$med_dur),
dataset = dt_source
)
return(res)
})
fsom_runtime_allcell <- rbindlist(fsom_runtime_allcell)
fsom_runtime <- rbind(
fsom_runtime_allcell,
fsom_runtime_supercell
)
fsom_runtime[, duration_seconds := as.numeric(duration_seconds)]
fsom_runtime[, log10_duration_seconds := log10(duration_seconds)]
fsom_runtime[, gamma := factor(gamma, levels = c("single cell", seq(5,50,5)))]ggplot(fsom_runtime, aes(x = gamma, y = duration_seconds, fill=dataset)) +
scale_fill_viridis(discrete = TRUE, alpha=0.6) +
geom_boxplot(outlier.size=0.2, lwd=0.2) +
theme_classic() +
facet_wrap(~dataset) +
scale_y_continuous(breaks = pretty_breaks(n=20)) +
labs(y = "Duration (seconds)", x = "Gamma", title = "Time taken to run FlowSOM clustering", fill="Dataset") +
theme(axis.text.x = element_text(angle = 45, hjust=1), legend.position="none")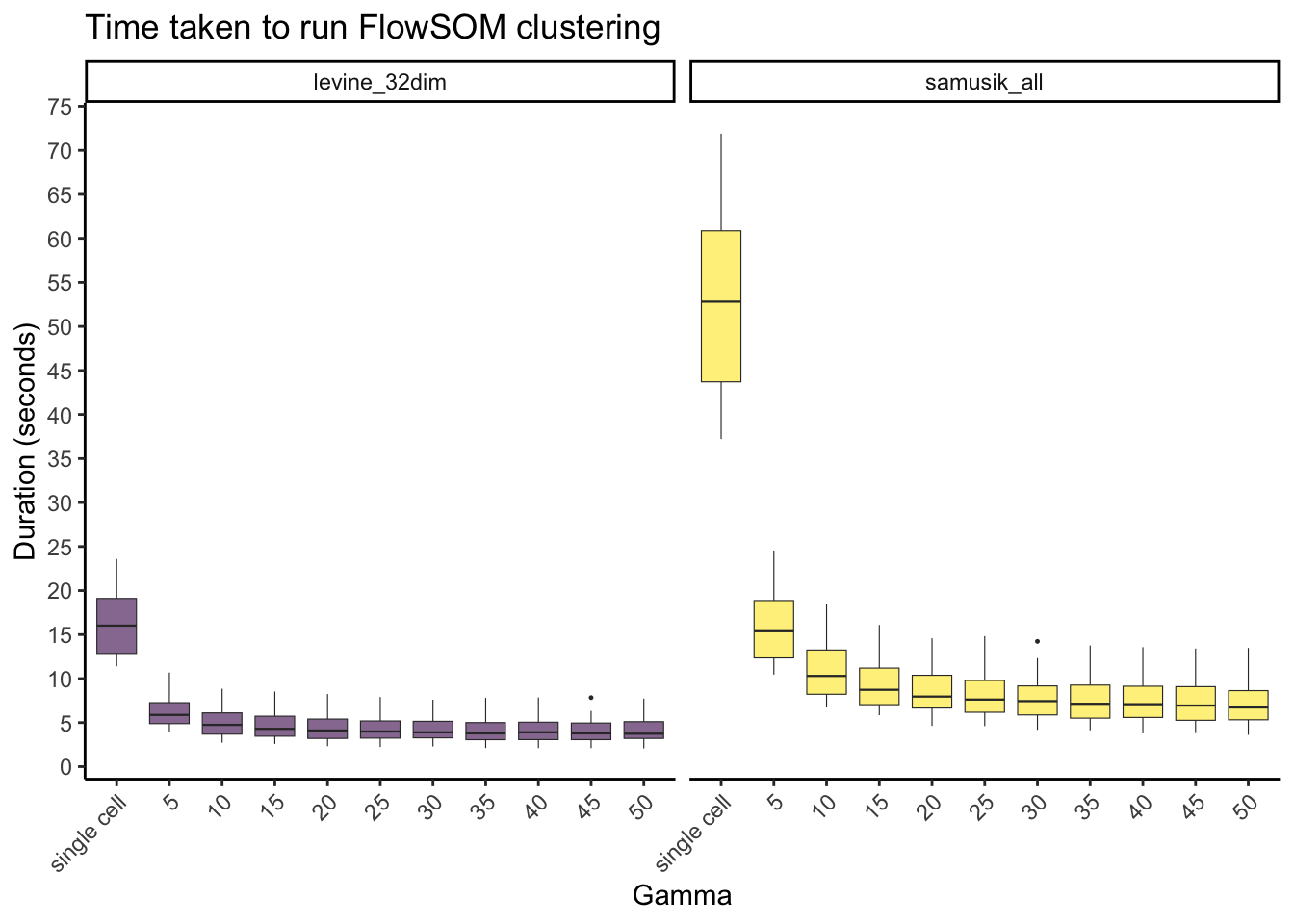
Label transfer
dur_files <- c("harmony_knn_runtime_singlecell.txt", "harmony_knn_runtime.txt",
"seurat_rPCA_singlecell_runtime.txt", "seurat_rPCA_runtime.txt")
label_trans <- sapply(dur_files, function(f) {
dt <- fread(paste0("output/label_transfer/", f))
return(dt$V3)
})
label_trans_dt <- data.table(
approah = names(label_trans),
duration_seconds = label_trans,
duration_minutes = label_trans/60
)
label_trans_dt approah duration_seconds duration_minutes
1: harmony_knn_runtime_singlecell.txt 323.237 5.387283
2: harmony_knn_runtime.txt 62.565 1.042750
3: seurat_rPCA_singlecell_runtime.txt 2197.163 36.619383
4: seurat_rPCA_runtime.txt 575.482 9.591367
sessionInfo()R version 4.2.3 (2023-03-15)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Monterey 12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] viridis_0.6.2 viridisLite_0.4.1 scales_1.2.1 ggplot2_3.4.1
[5] stringr_1.5.0 data.table_1.14.8 here_1.0.1 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.0 xfun_0.39 bslib_0.4.2 colorspace_2.1-0
[5] vctrs_0.5.2 generics_0.1.3 htmltools_0.5.4 yaml_2.3.7
[9] utf8_1.2.3 rlang_1.0.6 jquerylib_0.1.4 later_1.3.0
[13] pillar_1.8.1 glue_1.6.2 withr_2.5.0 RColorBrewer_1.1-3
[17] lifecycle_1.0.3 munsell_0.5.0 gtable_0.3.1 evaluate_0.20
[21] knitr_1.42 callr_3.7.3 fastmap_1.1.0 httpuv_1.6.9
[25] ps_1.7.2 fansi_1.0.4 highr_0.10 Rcpp_1.0.10
[29] promises_1.2.0.1 cachem_1.0.6 jsonlite_1.8.4 farver_2.1.1
[33] fs_1.6.1 gridExtra_2.3 digest_0.6.31 stringi_1.7.12
[37] processx_3.8.0 dplyr_1.1.0 getPass_0.2-2 rprojroot_2.0.3
[41] grid_4.2.3 cli_3.6.0 tools_4.2.3 magrittr_2.0.3
[45] sass_0.4.5 tibble_3.1.8 whisker_0.4.1 pkgconfig_2.0.3
[49] rmarkdown_2.20 httr_1.4.4 rstudioapi_0.14 R6_2.5.1
[53] git2r_0.31.0 compiler_4.2.3