How to Prepare Data for SuperCellCyto
Givanna Putri
Source:vignettes/how_to_prepare_data.Rmd
how_to_prepare_data.RmdPerforming Quality Control
Prior to creating supercells, it’s crucial to ensure that your dataset has undergone thorough quality control (QC). We want to retain only single, live cells and remove any debris, doublets, or dead cells. Additionally, it is also important to perform compensation to correct for fluorescence spillover (for Flow data) or to adjust for signal overlap or spillover between different metal isotopse (for Cytof data). A well-prepared dataset is key to obtaining reliable supercells from SuperCellCyto.
Several R packages are available for performing QC on cytometry data. Notable among these are PeacoQC, CATALYST, and CytoExploreR. These packages are well maintained and are continuously updated. To make sure that the information we provide do not quickly go out of date, we highly recommend you to consult the packages’ respective vignettes for detailed guidance on how to use them to QC your data.
In our manuscript, we used CytoExploreR to QC the
Oetjen_bcell flow cytometry data and CATALYST
to QC the Trussart_cytofruv Cytof data.
The specific scripts used can be found in Github:
-
b_cell_identification/gate_flow_data.RforOetjen_bcelldata. -
batch_correction/prepare_data.RforTrussart_cytofruvdata. These scripts were adapted from those used in the CytofRUV manuscript.
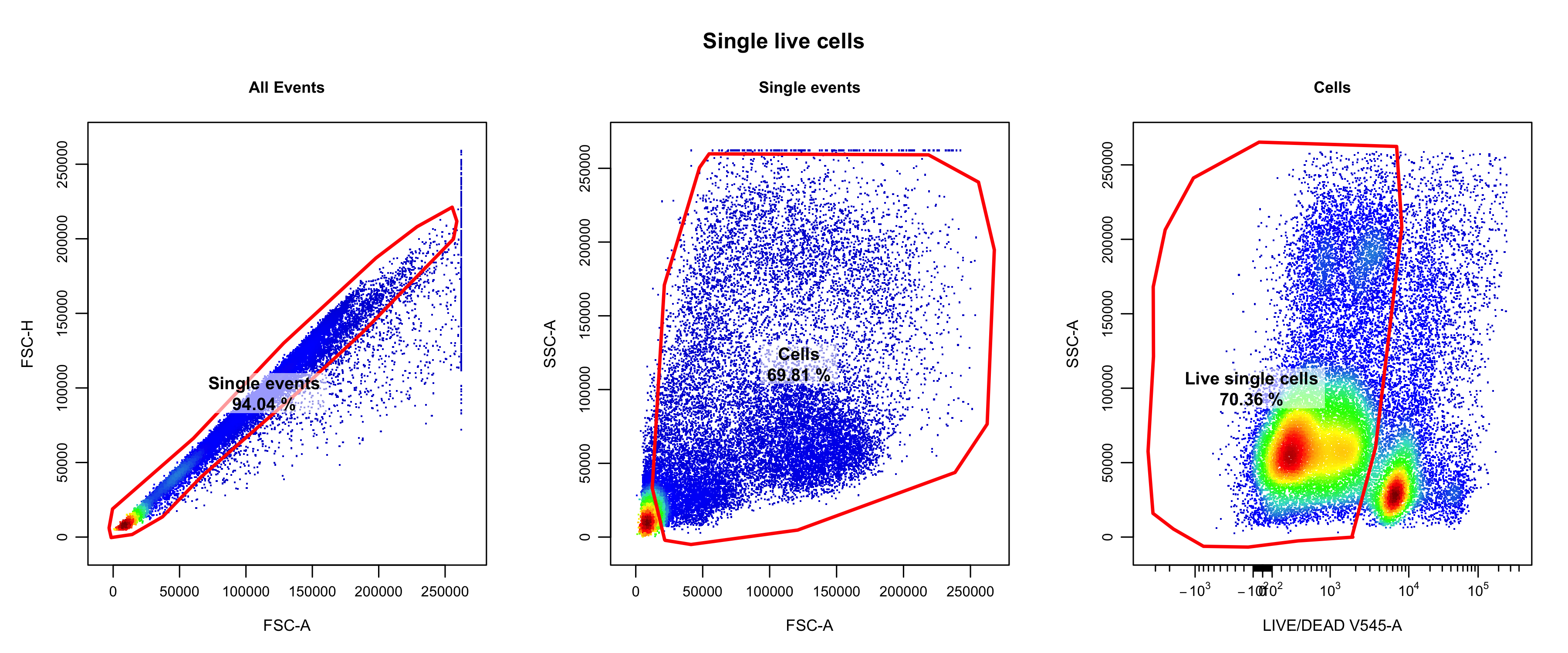
For Oetjen_bcell data, we used the following gating strategy post compensation:
- FSC-H and FSC-A to isolate only the single events. (Also check SSC-H vs SSC-A).
- FSC-A and SSC-A to remove debris.
- Live/Dead and SSC-A to isolate live cells.
The following is the resulting single live cells manually gated for
the Oetjen_bcell data.
knitr::include_graphics(
"figures/oetjen_bcell_single_live_cells.png",
error = FALSE
)
After completing the QC process, you will have clean data in either CSV or FCS file formats. The next section will guide you on how to load these files and proceed with preparing your data for SuperCellCyto.
Preparing FCS/CSV files for SuperCellCyto
To use SuperCellCyto, your input data must be formatted as a data.table
object. Briefly, data.table is an enhanced version of R
native data.frame object. It is a package that offers fast
processing of large data.frame.
Cell ID column
Additionally, each cell in your data.table must also
have a unique identifier. The purpose of this ID is to allow SuperCell
to uniquely identify each cell in the dataset. It will come in super
handy later when/if we need to work out which cells belong to which
supercells, i.e., when we need to expand the supercells out. Generally,
we will need to create this ID ourselves. Most dataset won’t come with
this ID already embedded in.
For this tutorial, we will call the column that denotes the cell ID cell_id. For your own dataset, you can name this column however you like, e.g., id, cell_identity, etc. Just make sure you note the column name as we will need it later to create supercells.
Sample column
Lastly, each cell in the data.table object must also be
associated with a sample. This information must be stored in a column
that we later on pass to the function that creates supercells.
Generally, sample here typically refers to the biological sample the
cell came from.
To create supercells, it is necessary to have this column in our dataset. This is to ensure that each supercell will only have cells from exactly one sample. In most cases, it does not make sense to mix cells from different biological samples in one supercell. Additionally (not as important), SuperCellCyto can process multiple samples in parallel, and for it to do that, it needs to know the sample information.
But what if we only have 1 biological sample in our dataset? It does not matter. We still need to have the sample column in our dataset. The only difference is that this column will only have 1 unique value.
You can name the column however we like, e.g., Samp, Cell_Samp, etc. For this tutorial, we will call the column sample. Just make sure you note the column name as we will need it later to create supercells.
Preparing CSV files
Loading CSV files into a data.table object is
straightforward. We can use the fread function from the
data.table package.
For this example, let’s load two CSV files containing subsampled data
from the Levine_32dim dataset we used in SuperCellCyto
manuscript. Each file represents a sample (H1 and H2), with the sample
name appended to the file name:
library(data.table)
csv_files <- system.file(
"extdata",
c("Levine_32dim_H1_sub.csv", "Levine_32dim_H2_sub.csv"),
package = "SuperCellCyto"
)
samples <- c("H1", "H2")
dat <- lapply(seq_len(length(samples)), function(i) {
csv_file <- csv_files[i]
sample <- samples[i]
dat_a_sample <- fread(csv_file)
dat_a_sample$sample <- sample
return(dat_a_sample)
})
dat <- rbindlist(dat)
dat[, cell_id := paste0("Cell_", seq_len(nrow(dat)))]
head(dat)
#> Time Cell_length DNA1 DNA2 CD45RA CD133 CD19
#> <num> <int> <num> <num> <num> <num> <num>
#> 1: 307428 27 169.91125 262.3192 2.338830 -0.1533398 -0.2056334
#> 2: 80712 13 50.91230 181.1320 2.129232 3.3563867 -0.1013980
#> 3: 284190 53 98.28069 187.2090 4.289627 0.5625419 12.2265682
#> 4: 324301 25 94.70678 258.6657 6.537235 0.3936968 37.9688492
#> 5: 253799 45 76.75870 128.8098 4.390123 0.3281800 0.3218679
#> 6: 313897 50 156.23492 254.4634 4.722823 1.5105748 -0.2037212
#> CD22 CD11b CD4 CD8 CD34 Flt3
#> <num> <num> <num> <num> <num> <num>
#> 1: -0.1972007 32.130402 0.78105438 -0.071934469 1.53498471 0.84833205
#> 2: 3.0564795 14.239288 0.53373063 -0.007943562 -0.09401329 -0.13234507
#> 3: 10.8176870 1.826709 1.30010796 -0.187664956 2.05419374 2.72891521
#> 4: 22.7653027 0.176247 -0.04688064 -0.075562298 3.77700782 0.01351853
#> 5: 2.2320855 1.343770 2.38195324 -0.120859891 -0.01110659 2.28398013
#> 6: 1.3113755 32.728638 0.33130702 -0.073869787 1.14790535 0.23651542
#> CD20 CXCR4 CD235ab CD45 CD123 CD321
#> <num> <num> <num> <num> <num> <num>
#> 1: -0.086762585 3.488938 0.8230118 313.8038 0.30909532 46.484669
#> 2: -0.042171009 1.364644 -0.1309417 207.2459 1.76594567 22.532978
#> 3: 15.341629982 9.303430 6.3413548 751.0563 0.05031190 10.463912
#> 4: 48.562511444 26.948063 0.2169415 588.5150 2.96119761 22.149944
#> 5: 3.535085201 2.498724 12.1216116 1007.4561 -0.18679842 5.777249
#> 6: -0.003814452 6.879488 4.4369431 918.5178 -0.03130747 24.542976
#> CD14 CD33 CD47 CD11c CD7 CD15
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.05072345 2.09802437 20.96871 20.7631855 -0.007966662 0.7279212
#> 2: -0.19256826 7.35230541 27.49848 15.1339817 -0.087256350 0.7187206
#> 3: 1.11993504 0.15872155 40.63973 4.6401038 -0.195279136 4.5712810
#> 4: -0.17467052 -0.18741834 7.64338 -0.1591413 0.604379535 1.1375968
#> 5: -0.20860019 0.04116142 30.60899 1.8959216 39.631023407 0.8820186
#> 6: 0.64118510 2.93926096 18.20175 34.3703728 -0.016664671 1.1723862
#> CD16 CD44 CD38 CD13 CD3 CD61
#> <num> <num> <num> <num> <num> <num>
#> 1: -0.03067662 95.71002 5.112477 5.10564327 0.5827813 -0.1684093
#> 2: 0.41139653 185.51929 7.478415 0.35808861 1.8861074 1.9233229
#> 3: -0.10192144 98.09428 43.535297 2.83275175 0.1868679 2.1032026
#> 4: 1.02131248 45.03027 3.396393 -0.09499715 0.8835158 5.2639704
#> 5: 0.28814790 81.63257 8.634666 0.80971009 728.0478516 1.9550949
#> 6: 2.46474814 250.00719 17.964558 0.38511425 1.4373373 9.9041309
#> CD117 CD49d HLA-DR CD64 CD41 Viability
#> <num> <num> <num> <num> <num> <num>
#> 1: -0.02967962 6.557199 112.4675446 6.9157209 0.08380865 1.726863
#> 2: -0.14122920 1.088500 12.2067947 30.7242870 7.75372791 3.712019
#> 3: 0.01776284 12.400333 174.9526672 0.4361930 1.83412540 13.274319
#> 4: 0.19813232 2.382564 3.4147604 0.9252884 -0.06184266 1.482011
#> 5: 3.34092522 1.395274 0.3110902 0.1227915 0.27702999 3.165646
#> 6: 0.46414420 14.136827 26.3479862 20.1809521 10.70872402 23.840090
#> file_number event_number sample cell_id
#> <int> <int> <char> <char>
#> 1: 94 257088 H1 Cell_1
#> 2: 94 80655 H1 Cell_2
#> 3: 94 241699 H1 Cell_3
#> 4: 94 268117 H1 Cell_4
#> 5: 94 221804 H1 Cell_5
#> 6: 94 261282 H1 Cell_6Let’s break down what we have done.
We specify the location of the csv files in csv_files
vector and their corresponding sample names in samples
vector. Levine_32dim_H1_sub.csv belongs to sample H1 while
Levine_32dim_H2_sub.csv belongs to sample H2.
We use lapply to simultaneously iterate over each
element in the csv_files and samples vector.
For each csv file and the corresponding sample, we read the csv file
into the variable dat_a_sample using fread
function. We then assign the sample id in a new column called
sample. As a result, we get a list dat
containing 2 data.table objects, 1 object per csv file.
We use rbindlist function from the
data.table package to merge list into one
data.table object.
We create a new column cell_id which gives each cell a
unique id such as Cell_1, Cell_2, etc.
Preparing FCS files
FCS files, commonly used in cytometry, require specific handling. You
can read in FCS files using the flowCore package available
from Bioconductor and convert it to a data.table
object.
Let’s load two small FCS files for the Anti-PD1 data from FlowRepository.
library(flowCore)
library(data.table)
fs <- read.flowSet(
path = system.file(
"extdata",
package = "SuperCellCyto"
),
pattern = "\\.fcs$"
)
dat_list <- lapply(seq_along(fs), function(i) {
df <- as.data.table(exprs(fs[[i]]))
# concatenate channel and marker name as column names
names(df) <- markernames(fs[[i]])
# add a column showing the filename
df$file_name <- sampleNames(fs)[i]
return(df)
})
# collate all the files into one
dat <- rbindlist(dat_list)
dat
#> 209Bi_CD11b 162Dy_CD11c 163Dy_CD7 166Er_CD209 167Er_CD38 151Eu_CD123
#> <num> <num> <num> <num> <num> <num>
#> 1: 2.596444 0.000000 24.233248 2.4271879 0.2497845 0.01359761
#> 2: 20.172476 29.508638 253.930161 2.0283859 63.4845619 0.00000000
#> 3: 224.771057 377.262421 20.755060 5.5264821 111.7728882 10.33144855
#> 4: 711.252808 192.961227 16.039482 2.7453365 13.4806919 1.71946943
#> 5: 435.131073 203.836838 5.983910 0.0000000 4.2271118 1.08193946
#> ---
#> 569: 337.737152 160.982407 1.724550 0.8164008 10.8179998 1.87148142
#> 570: 48.965187 3.454711 55.719162 0.7539494 20.6741104 0.00000000
#> 571: 326.666168 58.792435 4.358847 0.0000000 8.6697922 0.00000000
#> 572: 7.255682 0.000000 3.552515 0.0000000 2.9367595 0.43651727
#> 573: 349.350769 31.072611 1.050743 0.0000000 5.4545450 5.87748337
#> 153Eu_CD62L 152Gd_CD66b 154Gd_ICAM-1 155Gd_CD1c 156Gd_CD86 160Gd_CD14
#> <num> <num> <num> <num> <num> <num>
#> 1: 276.736237 0.0000000 6.3661394 0.72167069 0.0000000 0.000000
#> 2: 7.522890 0.0000000 16.6949806 0.00000000 0.9723848 2.370285
#> 3: 2.523962 0.0000000 480.9555969 2.87207913 109.6869812 41.803619
#> 4: 1.439798 0.0000000 125.3002014 0.00000000 5.1308088 19.226034
#> 5: 3.030660 0.0000000 54.1368217 0.03324713 23.3382854 14.204736
#> ---
#> 569: 0.000000 0.0000000 158.1088257 2.28826094 15.2002096 5.657256
#> 570: 3.414483 0.0000000 0.9569107 2.11766744 0.7849152 4.082072
#> 571: 0.000000 0.0000000 1.1182016 0.00000000 0.0000000 0.000000
#> 572: 2.556602 0.6154923 0.0000000 0.00000000 0.0000000 0.000000
#> 573: 3.925082 0.0000000 43.5083618 0.00000000 4.2216253 8.216695
#> 165Ho_CD16 191Ir_DNA1 193Ir_DNA2 175Lu_PD-L1 142Nd_CD19 146Nd_CD64
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.0000000 40.33384 76.70573 0.0000000 0.00000000 5.452389
#> 2: 126.4256592 55.11267 150.46278 3.4527724 0.00000000 1.922374
#> 3: 2.0556698 91.42932 161.59381 8.3969393 0.00000000 12.956987
#> 4: 0.0000000 75.32122 160.41399 0.1078656 0.00000000 12.841355
#> 5: 0.5523136 79.88800 142.12471 0.6882498 0.00000000 19.308508
#> ---
#> 569: 0.0000000 65.31769 148.84435 0.1064020 0.00000000 22.941177
#> 570: 51.3577194 65.16866 129.46790 3.5496852 0.36609724 2.852356
#> 571: 0.9855582 92.97505 151.65977 0.0000000 0.03485066 6.290884
#> 572: 0.0000000 65.07745 111.99138 0.5479890 0.63119560 0.000000
#> 573: 0.0000000 114.67633 191.50166 0.0000000 0.00000000 12.638340
#> 195Pt 196Pt 198Pt_Dead 147Sm_CD303 148Sm_CD34 149Sm_CD141
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.0000000 0.0000000 15.199163 0.0000000 1.2188276 2.482402
#> 2: 2.6318610 0.0000000 32.325874 0.0000000 0.2467884 0.000000
#> 3: 0.2854315 0.3602256 10.119688 0.7021694 7.2453775 17.055792
#> 4: 1.7483448 1.6977243 2.457203 0.0000000 1.1050161 6.811901
#> 5: 0.0000000 2.7839208 13.680239 0.8908378 0.0000000 18.400421
#> ---
#> 569: 0.0000000 0.0000000 16.751446 2.6801469 0.6307272 2.710867
#> 570: 0.3765990 0.0000000 20.695446 0.0000000 3.0734873 1.968072
#> 571: 0.1897834 0.0000000 16.076082 0.9444249 0.4878764 6.945232
#> 572: 0.0000000 0.0000000 23.279974 0.0000000 0.0000000 0.000000
#> 573: 0.0000000 4.0132923 12.463143 0.8065992 1.6200637 4.190233
#> 150Sm_CD61 169Tm_CD33 89Y_CD45 170Yb_CD3 173Yb_CD56 174Yb_HLA-DR
#> <num> <num> <num> <num> <num> <num>
#> 1: 65.015724 1.1326860 37.52760 9.0316811 0.00000000 6.807954
#> 2: 25.455738 0.1979908 225.64279 0.9135754 8.38446426 5.601559
#> 3: 75.056625 134.7255249 458.79022 12.2988071 10.31508160 403.328308
#> 4: 21.287003 58.7627487 387.78836 1.1870043 0.00000000 88.822212
#> 5: 26.795689 35.0521393 107.94828 0.3782265 5.60699320 104.159081
#> ---
#> 569: 36.163387 22.8519592 227.01973 2.7277973 0.02519703 188.378601
#> 570: 37.537052 3.3848262 48.12588 4.6816015 2.54012346 10.622149
#> 571: 16.739002 10.3474064 60.64515 0.6677586 0.00000000 15.465885
#> 572: 15.469506 0.0000000 11.47586 8.9600000 0.00000000 0.000000
#> 573: 8.587144 18.8689251 52.50787 0.6892268 1.88265443 13.542062
#> file_name
#> <char>
#> 1: Data23_Panel3_base_NR4_Patient9.fcs
#> 2: Data23_Panel3_base_NR4_Patient9.fcs
#> 3: Data23_Panel3_base_NR4_Patient9.fcs
#> 4: Data23_Panel3_base_NR4_Patient9.fcs
#> 5: Data23_Panel3_base_NR4_Patient9.fcs
#> ---
#> 569: Data23_Panel3_base_R5_Patient15.fcs
#> 570: Data23_Panel3_base_R5_Patient15.fcs
#> 571: Data23_Panel3_base_R5_Patient15.fcs
#> 572: Data23_Panel3_base_R5_Patient15.fcs
#> 573: Data23_Panel3_base_R5_Patient15.fcsThe code above used flowCore’s read.flowSet
function to first read FCS files into a flowSet object.
lapply and rbindlist is then used to
convert it to one data.table object containing data from
all FCS files.
The FCS files belong to two different patients, patient 9 and 15. We
shall use that as the sample ID. To make sure that we correctly map the
filenames to the patients, we will first create a new
data.table object containing the mapping of FileName and
the sample name, and then using merge.data.table to add
them into our data.table object.
We will also to create a new column cell_id which gives
each cell a unique id such as Cell_1, Cell_2,
etc.
sample_info <- data.table(
sample = c("patient9", "patient15"),
file_name = c(
"Data23_Panel3_base_NR4_Patient9.fcs",
"Data23_Panel3_base_R5_Patient15.fcs"
)
)
dat <- merge.data.table(
x = dat,
y = sample_info,
by = "file_name"
)
dat[, cell_id := paste0("Cell_", seq_len(nrow(dat)))]With CSV and FCS files loaded as data.table objects, the next step is to transform the data appropriately for SuperCellCyto.
Data Transformation
Before using SuperCellCyto, it’s essential to apply appropriate data transformations.
A common method for data transformation in cytometry is the arcsinh transformation, an inverse hyperbolic arcsinh transformation. The transformation requires specifying a cofactor, which affects the representation of the low-end data. Typically, a cofactor of 5 is used for Cytof data and 150 for Flow data. This vignette will focus on the transformation process rather than cofactor selection.
We’ll use the Levine_32dim dataset loaded earlier from
CSV files.
First, we need to select the markers to be transformed. Usually, all markers should be transformed for SuperCellCyto. However, you can choose to exclude specific markers if needed:
markers <- c(
"209Bi_CD11b", "162Dy_CD11c", "163Dy_CD7", "166Er_CD209", "167Er_CD38",
"151Eu_CD123", "153Eu_CD62L", "152Gd_CD66b", "154Gd_ICAM-1", "155Gd_CD1c",
"156Gd_CD86", "160Gd_CD14", "165Ho_CD16", "191Ir_DNA1", "193Ir_DNA2",
"175Lu_PD-L1", "142Nd_CD19", "146Nd_CD64", "195Pt", "196Pt",
"198Pt_Dead", "147Sm_CD303", "148Sm_CD34", "149Sm_CD141", "150Sm_CD61",
"169Tm_CD33", "89Y_CD45", "170Yb_CD3", "173Yb_CD56", "174Yb_HLA-DR"
)For transformation, we’ll use a cofactor of 5 and apply the arcsinh transformation.
new_cols <- paste0(markers, "_asinh")
cf <- 5
dat[, (new_cols) := lapply(.SD, function(x) asinh(x / cf)), .SDcols = markers]After transformation, new columns with “_asinh” appended indicate the transformed markers.
head(dat)
#> Key: <file_name>
#> file_name 209Bi_CD11b 162Dy_CD11c 163Dy_CD7
#> <char> <num> <num> <num>
#> 1: Data23_Panel3_base_NR4_Patient9.fcs 2.596444 0.00000 24.233248
#> 2: Data23_Panel3_base_NR4_Patient9.fcs 20.172476 29.50864 253.930161
#> 3: Data23_Panel3_base_NR4_Patient9.fcs 224.771057 377.26242 20.755060
#> 4: Data23_Panel3_base_NR4_Patient9.fcs 711.252808 192.96123 16.039482
#> 5: Data23_Panel3_base_NR4_Patient9.fcs 435.131073 203.83684 5.983910
#> 6: Data23_Panel3_base_NR4_Patient9.fcs 182.881317 244.65285 5.262897
#> 166Er_CD209 167Er_CD38 151Eu_CD123 153Eu_CD62L 152Gd_CD66b 154Gd_ICAM-1
#> <num> <num> <num> <num> <num> <num>
#> 1: 2.427188 0.2497845 0.01359761 276.736237 0 6.366139
#> 2: 2.028386 63.4845619 0.00000000 7.522890 0 16.694981
#> 3: 5.526482 111.7728882 10.33144855 2.523962 0 480.955597
#> 4: 2.745337 13.4806919 1.71946943 1.439798 0 125.300201
#> 5: 0.000000 4.2271118 1.08193946 3.030660 0 54.136822
#> 6: 1.548412 34.4840889 1.36238360 0.000000 0 13.646034
#> 155Gd_CD1c 156Gd_CD86 160Gd_CD14 165Ho_CD16 191Ir_DNA1 193Ir_DNA2
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.72167069 0.0000000 0.000000 0.0000000 40.33384 76.70573
#> 2: 0.00000000 0.9723848 2.370285 126.4256592 55.11267 150.46278
#> 3: 2.87207913 109.6869812 41.803619 2.0556698 91.42932 161.59381
#> 4: 0.00000000 5.1308088 19.226034 0.0000000 75.32122 160.41399
#> 5: 0.03324713 23.3382854 14.204736 0.5523136 79.88800 142.12471
#> 6: 4.62103748 11.6817465 2.560833 0.0000000 56.35513 93.84315
#> 175Lu_PD-L1 142Nd_CD19 146Nd_CD64 195Pt 196Pt 198Pt_Dead 147Sm_CD303
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 0.0000000 0 5.452389 0.0000000 0.0000000 15.199163 0.0000000
#> 2: 3.4527724 0 1.922374 2.6318610 0.0000000 32.325874 0.0000000
#> 3: 8.3969393 0 12.956987 0.2854315 0.3602256 10.119688 0.7021694
#> 4: 0.1078656 0 12.841355 1.7483448 1.6977243 2.457203 0.0000000
#> 5: 0.6882498 0 19.308508 0.0000000 2.7839208 13.680239 0.8908378
#> 6: 5.6380033 0 8.388703 0.0000000 0.0000000 3.202339 0.7597103
#> 148Sm_CD34 149Sm_CD141 150Sm_CD61 169Tm_CD33 89Y_CD45 170Yb_CD3 173Yb_CD56
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 1.2188276 2.482402 65.01572 1.1326860 37.5276 9.0316811 0.000000
#> 2: 0.2467884 0.000000 25.45574 0.1979908 225.6428 0.9135754 8.384464
#> 3: 7.2453775 17.055792 75.05663 134.7255249 458.7902 12.2988071 10.315082
#> 4: 1.1050161 6.811901 21.28700 58.7627487 387.7884 1.1870043 0.000000
#> 5: 0.0000000 18.400421 26.79569 35.0521393 107.9483 0.3782265 5.606993
#> 6: 4.0056577 2.323631 12.82973 55.7610817 444.4799 0.6113114 1.556887
#> 174Yb_HLA-DR sample cell_id 209Bi_CD11b_asinh 162Dy_CD11c_asinh
#> <num> <char> <char> <num> <num>
#> 1: 6.807954 patient9 Cell_1 0.4983974 0.000000
#> 2: 5.601559 patient9 Cell_2 2.1030451 2.475494
#> 3: 403.328308 patient9 Cell_3 4.4989153 5.016694
#> 4: 88.822212 patient9 Cell_4 5.6507496 4.346366
#> 5: 104.159081 patient9 Cell_5 5.1593896 4.401180
#> 6: 145.182190 patient9 Cell_6 4.2927335 4.583654
#> 163Dy_CD7_asinh 166Er_CD209_asinh 167Er_CD38_asinh 151Eu_CD123_asinh
#> <num> <num> <num> <num>
#> 1: 2.2819117 0.4681490 0.04993614 0.002719519
#> 2: 4.6208655 0.3953013 3.23605319 0.000000000
#> 3: 2.1307022 0.9539050 3.80067819 1.472894089
#> 4: 1.8822162 0.5246630 1.71770981 0.337452783
#> 5: 1.0139113 0.0000000 0.76774561 0.214733831
#> 6: 0.9180684 0.3049346 2.62942218 0.269213018
#> 153Eu_CD62L_asinh 152Gd_CD66b_asinh 154Gd_ICAM-1_asinh 155Gd_CD1c_asinh
#> <num> <num> <num> <num>
#> 1: 4.7068557 0 1.062022 0.143837642
#> 2: 1.1972999 0 1.920522 0.000000000
#> 3: 0.4854943 0 5.259511 0.546763229
#> 4: 0.2841215 0 3.914820 0.000000000
#> 5: 0.5740759 0 3.077350 0.006649377
#> 6: 0.0000000 0 1.729148 0.826752274
#> 156Gd_CD86_asinh 160Gd_CD14_asinh 165Ho_CD16_asinh 191Ir_DNA1_asinh
#> <num> <num> <num> <num>
#> 1: 0.0000000 0.0000000 0.0000000 2.784720
#> 2: 0.1932715 0.4578881 3.9237545 3.095140
#> 3: 3.7818590 2.8202496 0.4003530 3.600022
#> 4: 0.8997523 2.0564695 0.0000000 3.406571
#> 5: 2.2450865 1.7669123 0.1102393 3.465313
#> 6: 1.5846764 0.4920673 0.0000000 3.117345
#> 193Ir_DNA2_asinh 175Lu_PD-L1_asinh 142Nd_CD19_asinh 146Nd_CD64_asinh
#> <num> <num> <num> <num>
#> 1: 3.424746 0.00000000 0 0.9439264
#> 2: 4.097701 0.64491132 0 0.3755822
#> 3: 4.169034 1.29032235 0 1.6806506
#> 4: 4.161710 0.02157144 0 1.6722922
#> 5: 4.040723 0.13721893 0 2.0606128
#> 6: 3.626043 0.96878631 0 1.2894793
#> 195Pt_asinh 196Pt_asinh 198Pt_Dead_asinh 147Sm_CD303_asinh 148Sm_CD34_asinh
#> <num> <num> <num> <num> <num>
#> 1: 0.00000000 0.00000000 1.8309680 0.0000000 0.24141372
#> 2: 0.50467458 0.00000000 2.5655054 0.0000000 0.04933766
#> 3: 0.05705535 0.07198295 1.4542897 0.1399763 1.16617935
#> 4: 0.34290908 0.33333742 0.4735430 0.0000000 0.21924260
#> 5: 0.00000000 0.53141631 1.7314988 0.1772382 0.00000000
#> 6: 0.00000000 0.00000000 0.6032147 0.1513634 0.73355159
#> 149Sm_CD141_asinh 150Sm_CD61_asinh 169Tm_CD33_asinh 89Y_CD45_asinh
#> <num> <num> <num> <num>
#> 1: 0.4780616 3.259814 0.22464301 2.713195
#> 2: 0.0000000 2.330159 0.03958781 4.502785
#> 3: 1.9410230 3.403060 3.98729298 5.212332
#> 4: 1.1159197 2.155322 3.15902249 5.044211
#> 5: 2.0140514 2.380543 2.64559441 3.765897
#> 6: 0.4494416 1.671448 3.10678950 5.180646
#> 170Yb_CD3_asinh 173Yb_CD56_asinh 174Yb_HLA-DR_asinh
#> <num> <num> <num>
#> 1: 1.35351374 0.0000000 1.1154525
#> 2: 0.18171341 1.2890452 0.9639413
#> 3: 1.63218235 1.4714672 5.0834986
#> 4: 0.23522564 0.0000000 3.5711373
#> 5: 0.07557334 0.9646649 3.7302042
#> 6: 0.12195971 0.3065534 4.0619951With your data now transformed, you’re ready to create supercells using SuperCellCyto. Please refer to How to create supercells vignette for detailed instructions.
Session information
sessionInfo()
#> R version 4.5.1 (2025-06-13)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] flowCore_2.20.0 data.table_1.17.8 BiocStyle_2.36.0
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.50 rlang_1.1.6
#> [4] xfun_0.53 png_0.1-8 generics_0.1.4
#> [7] textshaping_1.0.4 jsonlite_2.0.0 RProtoBufLib_2.20.0
#> [10] S4Vectors_0.46.0 htmltools_0.5.8.1 stats4_4.5.1
#> [13] ragg_1.5.0 sass_0.4.10 Biobase_2.68.0
#> [16] rmarkdown_2.30 evaluate_1.0.5 jquerylib_0.1.4
#> [19] fastmap_1.2.0 yaml_2.3.10 lifecycle_1.0.4
#> [22] bookdown_0.45 BiocManager_1.30.26 compiler_4.5.1
#> [25] fs_1.6.6 htmlwidgets_1.6.4 systemfonts_1.3.1
#> [28] digest_0.6.37 R6_2.6.1 cytolib_2.20.0
#> [31] bslib_0.9.0 tools_4.5.1 matrixStats_1.5.0
#> [34] BiocGenerics_0.54.1 pkgdown_2.1.3 cachem_1.1.0
#> [37] desc_1.4.3