Interoperability with SingleCellExperiment
Givanna Putri
Source:vignettes/interoperability_with_sce.Rmd
interoperability_with_sce.RmdIntroduction
This vignette demonstrates how to integrate SuperCellCyto results with cytometry data stored in SingleCellExperiment (SCE) objects, and how to analyse SuperCellCyto output using Bioconductor packages that take SCE objects as input.
We use a subsampled Levine_32dim dataset stored in a csv file to illustrate how to create supercells and conduct downstream analyses.
library(SuperCellCyto)
library(SingleCellExperiment)
library(scran)
library(BiocSingular)
library(scater)
library(bluster)
library(data.table)Preparing SCE object
We first create SCE object for our data.
dt <- fread(
system.file(
"extdata",
"Levine_32dim_subsampledPopulation.csv",
package = "SuperCellCyto"
)
)
head(dt)
#> population_id sample Time Cell_length DNA1 DNA2 CD45RA
#> <char> <char> <num> <int> <num> <num> <num>
#> 1: Basophils H1 328676 22 142.5924 322.3459 3.4128168
#> 2: Basophils H1 196440 29 130.1518 226.9475 -0.2144381
#> 3: Basophils H1 96186 22 71.5029 185.1046 4.3640466
#> 4: Basophils H1 52765 14 146.2525 275.0873 0.1746820
#> 5: Basophils H1 145738 56 101.4113 213.4994 2.0969417
#> 6: Basophils H1 93176 31 169.6428 302.5010 4.8693080
#> CD133 CD19 CD22 CD11b CD4 CD8
#> <num> <num> <num> <num> <num> <num>
#> 1: 1.62150514 0.029806657 -0.1518671 32.59288406 -0.003470583 -0.09476787
#> 2: -0.18932946 2.887274742 0.2699839 6.26368523 1.713465929 0.39009288
#> 3: -0.17672275 1.302536368 2.0841556 6.47213840 -0.217427284 -0.18387035
#> 4: 1.19722629 0.248987988 0.3383384 11.69494057 -0.189887092 -0.20784403
#> 5: -0.03515208 -0.191935569 -0.1749713 3.07195950 -0.114936128 -0.03475836
#> 6: -0.00656661 -0.008021533 -0.1996537 -0.05626484 2.429884672 -0.06190102
#> CD34 Flt3 CD20 CXCR4 CD235ab CD45 CD123
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 2.615368 0.4601151 0.2572394 6.2098589 0.21614286 388.97647 192.41806
#> 2: 1.314342 0.2938706 0.5782985 5.2307558 2.11593270 94.02493 310.79724
#> 3: 2.028696 4.6148148 -0.2100324 4.2273989 0.03399638 429.00729 295.18945
#> 4: 2.735096 0.4119383 1.6195409 0.4746386 4.64794445 247.72487 190.54358
#> 5: 1.799894 0.4545839 1.9519267 1.6836491 3.88455939 130.46397 173.23378
#> 6: 1.266070 2.0304530 0.3969584 2.5239871 4.06032562 118.22239 81.85568
#> CD321 CD14 CD33 CD47 CD11c CD7
#> <num> <num> <num> <num> <num> <num>
#> 1: 27.802330 -0.2088212 -0.08460895 51.82586 -0.1124191 -0.019866187
#> 2: 18.948410 0.1767059 -0.15765716 113.24429 0.6747438 -0.133409590
#> 3: 33.604641 -0.1651236 0.24777852 71.76296 0.1974210 -0.122436650
#> 4: 18.965446 -0.1292862 -0.18352161 66.36990 1.5187590 -0.067056000
#> 5: 31.202065 1.6112406 3.75688243 55.84665 8.8415623 0.783188105
#> 6: 5.323217 -0.1656112 0.13076653 58.75228 0.8378633 -0.008319224
#> CD15 CD16 CD44 CD38 CD13 CD3 CD61
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: -0.21626113 -0.1456568 32.07421 18.99353 8.2386065 1.15568256 3.2522867
#> 2: 2.74286819 1.6651863 183.19774 87.07681 3.7393653 0.80448067 2.6631687
#> 3: -0.02418897 -0.1232872 282.82016 197.23349 3.2029536 -0.22213262 7.2179780
#> 4: 0.67197126 -0.1876704 213.06563 127.52438 3.1244855 -0.02746344 4.4325719
#> 5: 6.17448807 3.4632318 172.03450 96.92493 3.5525737 2.00662255 2.2441761
#> 6: 3.17291641 0.4419518 114.73201 14.47360 0.4973633 -0.14863963 0.3224087
#> CD117 CD49d HLA-DR CD64 CD41 Viability
#> <num> <num> <num> <num> <num> <num>
#> 1: 0.39386749 1.39197588 2.9646072 0.7340060 5.73157263 0.22928734
#> 2: 0.12489000 4.06977797 1.0504816 1.6814929 2.06527948 10.62689114
#> 3: -0.09931306 -0.08391164 0.5018697 0.4667510 4.74700022 0.03840053
#> 4: -0.13148299 4.59189224 1.3115046 -0.1710277 6.59923792 0.09172521
#> 5: 1.01612949 9.79839325 3.2883773 1.9836804 0.60133815 9.15380859
#> 6: -0.03559279 15.46671772 12.5567837 0.8468229 -0.09069601 2.43577790
#> file_number event_number cell_id
#> <int> <int> <char>
#> 1: 94 270897 cell_1
#> 2: 94 183099 cell_2
#> 3: 94 99497 cell_3
#> 4: 94 46335 cell_4
#> 5: 94 146404 cell_5
#> 6: 94 95891 cell_6The dataset contains 32 markers (features) and have some metadata
columns like population_id, sample, and
cell_id. We need to make sure that the metadata columns are
not included in the expression matrix when creating the SCE object.
exprs_mat <- t(
as.matrix(dt[, !c("population_id", "sample", "cell_id"), with = FALSE])
)
sce <- SingleCellExperiment(
assays = list(counts = exprs_mat),
colData = dt[, c("population_id", "sample", "cell_id"), with = FALSE]
)
# assign cell ids as column names
colnames(sce) <- dt$cell_id
sce
#> class: SingleCellExperiment
#> dim: 39 1500
#> metadata(0):
#> assays(1): counts
#> rownames(39): Time Cell_length ... file_number event_number
#> rowData names(0):
#> colnames(1500): cell_1 cell_2 ... cell_1499 cell_1500
#> colData names(3): population_id sample cell_id
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):The data is stored in the counts assay. We will subset
it to include only markers we need to perform downstream analysis,
transform it using arcsinh transformation, and store the transformed
data in the logcounts assay.
markers <- c(
"CD45RA", "CD133", "CD19", "CD22", "CD11b", "CD4", "CD8",
"CD34", "Flt3", "CD20", "CXCR4", "CD235ab", "CD45", "CD123", "CD321",
"CD14", "CD33", "CD47", "CD11c", "CD7", "CD15", "CD16", "CD44", "CD38",
"CD13", "CD3", "CD61", "CD117",
"CD49d", "HLA-DR", "CD64", "CD41"
)
# keep only the relevant markers
sce <- sce[markers, ]
# to store arcsinh transformed data
exprs(sce) <- asinh(counts(sce) / 5)
sce
#> class: SingleCellExperiment
#> dim: 32 1500
#> metadata(0):
#> assays(2): counts logcounts
#> rownames(32): CD45RA CD133 ... CD64 CD41
#> rowData names(0):
#> colnames(1500): cell_1 cell_2 ... cell_1499 cell_1500
#> colData names(3): population_id sample cell_id
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):Run SuperCellCyto
SuperCellCyto requires input data in a data.table
format. Therefore, we need to extract the arcsinh-transformed data into
a data.table object, and add the sample information and IDs
of the cells.
Note that SCE typically stores cells as columns and features as rows.
SuperCellCyto, conversely, requires cells as rows and features as
columns, a format typical for cytometry data where we typically have
more cells than features. Hence, we will transpose the extracted data
accordingly when creating the data.table object.
dt <- data.table(t(exprs(sce)))
dt$sample <- colData(sce)$sample
dt$cell_id <- colnames(sce)
supercells <- runSuperCellCyto(
dt = dt,
markers = markers,
sample_colname = "sample",
cell_id_colname = "cell_id",
gam = 5
)
head(supercells$supercell_expression_matrix)
#> CD45RA CD133 CD19 CD22 CD11b CD4 CD8
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 0.7567786 0.2094107 0.07620974 0.009254529 0.3511375 0.78500942 0.8687925975
#> 2: 1.1482748 0.6268151 0.40876592 0.091766210 0.2766514 0.09603202 1.3321355589
#> 3: 0.1700757 0.1339944 0.07228738 0.101802043 0.1040339 0.18761509 0.1132434207
#> 4: 0.8968968 0.2921571 2.22313786 0.450342888 0.4193913 0.23917011 0.1332567931
#> 5: 0.1476499 0.1904578 0.11589638 0.106288009 0.2489399 0.52420384 0.4813924176
#> 6: 0.3801373 0.2772991 0.68028804 0.237641653 0.2007602 0.04097948 0.0001923242
#> CD34 Flt3 CD20 CXCR4 CD235ab CD45 CD123
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 0.11526045 0.5488327 0.041534679 1.2172962 0.6303494 5.788989 0.070171026
#> 2: 0.12708141 0.0793773 0.053358629 0.3920845 0.5637494 5.785661 0.084488379
#> 3: 3.23152402 0.1749861 0.008712508 0.5421237 0.1212147 3.416994 0.066197760
#> 4: 0.30587619 0.5787963 0.136006539 1.3302008 0.3211722 4.369223 0.698444542
#> 5: 0.05301752 0.3468969 0.092965825 0.2609136 0.5636011 5.684187 -0.003512615
#> 6: 2.13054119 0.1427986 0.005927587 0.5651623 0.1386637 3.051677 0.143230069
#> CD321 CD14 CD33 CD47 CD11c CD7 CD15
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 1.500236 0.04154518 0.06746186 3.221072 0.2123768 2.28170702 0.2050279
#> 2: 1.656612 0.08216914 0.12675445 2.043989 0.1105668 2.43975076 0.2104722
#> 3: 2.803258 0.02104697 0.17107591 2.657387 0.3190474 0.03814356 0.2649500
#> 4: 2.654545 0.01727268 0.24770161 4.576991 0.4200163 0.09815344 0.3564437
#> 5: 1.466286 0.08933425 0.02555677 2.352106 0.1591512 2.55868425 0.1185639
#> 6: 2.040685 0.09609030 0.35279157 3.578527 0.2416795 0.02281908 0.5117449
#> CD16 CD44 CD38 CD13 CD3 CD61 CD117
#> <num> <num> <num> <num> <num> <num> <num>
#> 1: 0.03168278 2.925953 0.6649237 0.09905437 5.10948135 0.05358984 0.281380801
#> 2: 0.10139879 4.354774 1.1550810 0.22855225 0.58614888 0.16057716 0.003634178
#> 3: 0.04358262 2.895200 0.9998574 0.31835180 0.15497706 0.05665786 0.993750340
#> 4: 0.35695241 5.021503 6.2330245 0.64945662 0.17109374 0.18993344 0.082566733
#> 5: 0.08155028 2.924965 0.3143248 0.09534336 4.89098214 0.22921411 0.059164421
#> 6: 0.06652319 2.830727 3.7236044 0.71869068 0.09991223 0.28525846 0.111576001
#> CD49d HLA-DR CD64 CD41 sample SuperCellId
#> <num> <num> <num> <num> <char> <char>
#> 1: 0.3529696 0.1407416 0.09377686 0.07614376 H1 SuperCell_1_Sample_H1
#> 2: 0.8440716 0.2467888 0.18515576 0.28544083 H1 SuperCell_2_Sample_H1
#> 3: 1.0233080 1.6318626 0.25253230 0.04277276 H1 SuperCell_3_Sample_H1
#> 4: 1.5830615 0.0820442 0.03792944 0.16956875 H1 SuperCell_4_Sample_H1
#> 5: 0.3508159 0.1748238 0.11055986 0.04522353 H1 SuperCell_5_Sample_H1
#> 6: 1.6866826 3.0233062 0.13634514 0.40852578 H1 SuperCell_6_Sample_H1We can now embed the supercell ID in the colData of our
SCE object.
colData(sce)$supercell_id <- factor(supercells$supercell_cell_map$SuperCellID)
head(colData(sce))
#> DataFrame with 6 rows and 4 columns
#> population_id sample cell_id supercell_id
#> <character> <character> <character> <factor>
#> cell_1 Basophils H1 cell_1 SuperCell_22_Sample_H1
#> cell_2 Basophils H1 cell_2 SuperCell_50_Sample_H1
#> cell_3 Basophils H1 cell_3 SuperCell_28_Sample_H1
#> cell_4 Basophils H1 cell_4 SuperCell_141_Sample_H1
#> cell_5 Basophils H1 cell_5 SuperCell_141_Sample_H1
#> cell_6 Basophils H1 cell_6 SuperCell_99_Sample_H1Analyse Supercells as SCE object
As the number of supercells is less than the number of cells in our SCE object, we store the supercell expression matrix as a separate SCE object. This then allows us to use Bioconductor packages to analyse our supercells.
supercell_sce <- SingleCellExperiment(
list(logcounts = t(
supercells$supercell_expression_matrix[, markers, with = FALSE]
)),
colData = DataFrame(
SuperCellId = supercells$supercell_expression_matrix$SuperCellId,
sample = supercells$supercell_expression_matrix$sample
)
)
colnames(supercell_sce) <- colData(supercell_sce)$SuperCellId
supercell_sce
#> class: SingleCellExperiment
#> dim: 32 300
#> metadata(0):
#> assays(1): logcounts
#> rownames(32): CD45RA CD133 ... CD64 CD41
#> rowData names(0):
#> colnames(300): SuperCell_1_Sample_H1 SuperCell_2_Sample_H1 ...
#> SuperCell_149_Sample_H2 SuperCell_150_Sample_H2
#> colData names(2): SuperCellId sample
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):The code above essentially transpose the supercell expression matrix,
making supercells columns and markers rows, and store it in the
logcounts assay of our new SCE object. We also populate the
colData with SuperCellId and the sample name for each
supercell.
With the supercell expression matrix now in an SCE format, we can perform downstream analyses such as clustering and and drawing UMAP plots using Bioconductor packages.
set.seed(42)
supercell_sce <- fixedPCA(
supercell_sce,
rank = 10,
subset.row = NULL,
BSPARAM = RandomParam()
)
supercell_sce <- runUMAP(supercell_sce, dimred = "PCA")
clusters <- clusterCells(
supercell_sce, use.dimred = "PCA",
BLUSPARAM = SNNGraphParam(cluster.fun = "leiden")
)
colLabels(supercell_sce) <- clusters
plotReducedDim(supercell_sce, dimred = "UMAP", colour_by = "label")
Any functions which operate on SCE object should work. For example,
we can use plotExpression in scater package to
create violin plots of the markers against clusters.
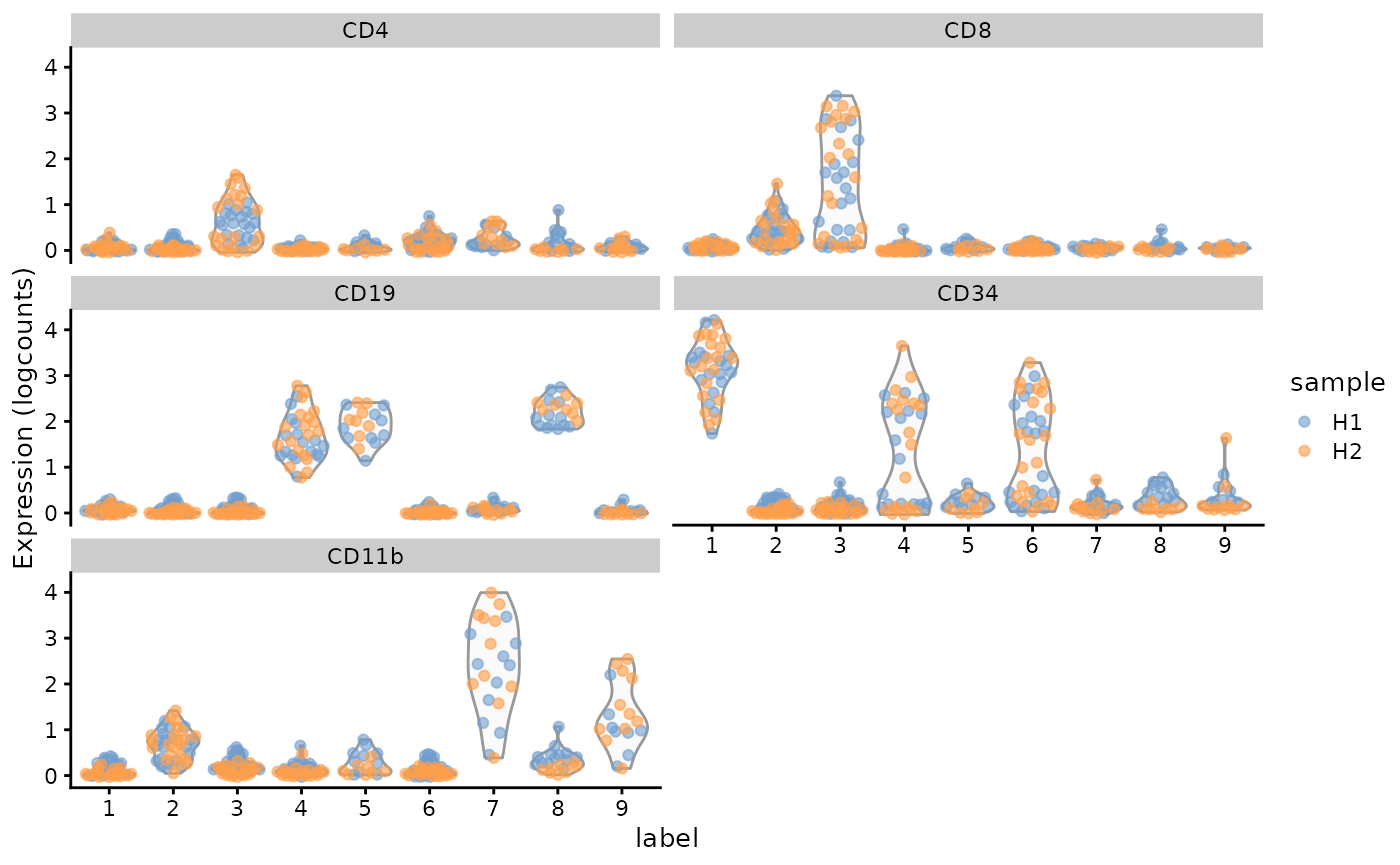
Note, the y-axis says “logcounts”, but the data is actually arcsinh transformed, not log transformed.
plotExpression(
supercell_sce, c("CD4", "CD8", "CD19", "CD34", "CD11b"),
x = "label", colour_by = "sample"
)
Transfer information from supercells SCE object to single cell SCE object
To transfer analysis results (e.g., clusters) from the supercell SCE object back to the single-cell SCE object, we need to do some data wrangling. It is vital to ensure that the order of the analysis results (e.g., clusters) aligns with the cell order in the SCE object.
Using the cluster information as an example, we need to first extract
the colData of the SCE objects into two separate
data.table objects. We then use
merge.data.table to match and merge them using the
supercell ID as the common identifiers. Make sure you set the
sort parameter to FALSE and set x to the
colData of your single cell SCE object. This ensures that
the order of the resulting data.table aligns with the order
of the colData of our single-cell SCE object.
cell_id_sce <- data.table(as.data.frame(colData(sce)))
supercell_cluster <- data.table(as.data.frame(colData(supercell_sce)))
cell_id_sce_with_clusters <- merge.data.table(
x = cell_id_sce,
y = supercell_cluster,
by.x = "supercell_id",
by.y = "SuperCellId",
sort = FALSE
)Finally, we can then add the cluster assignment as a column in the
colData of our single-cell SCE object.
colData(sce)$cluster <- cell_id_sce_with_clusters$labelVisualise them as UMAP plot.
sce <- fixedPCA(sce, rank = 10, subset.row = NULL, BSPARAM = RandomParam())
sce <- runUMAP(sce, dimred = "PCA")
plotReducedDim(sce, dimred = "UMAP", colour_by = "cluster")
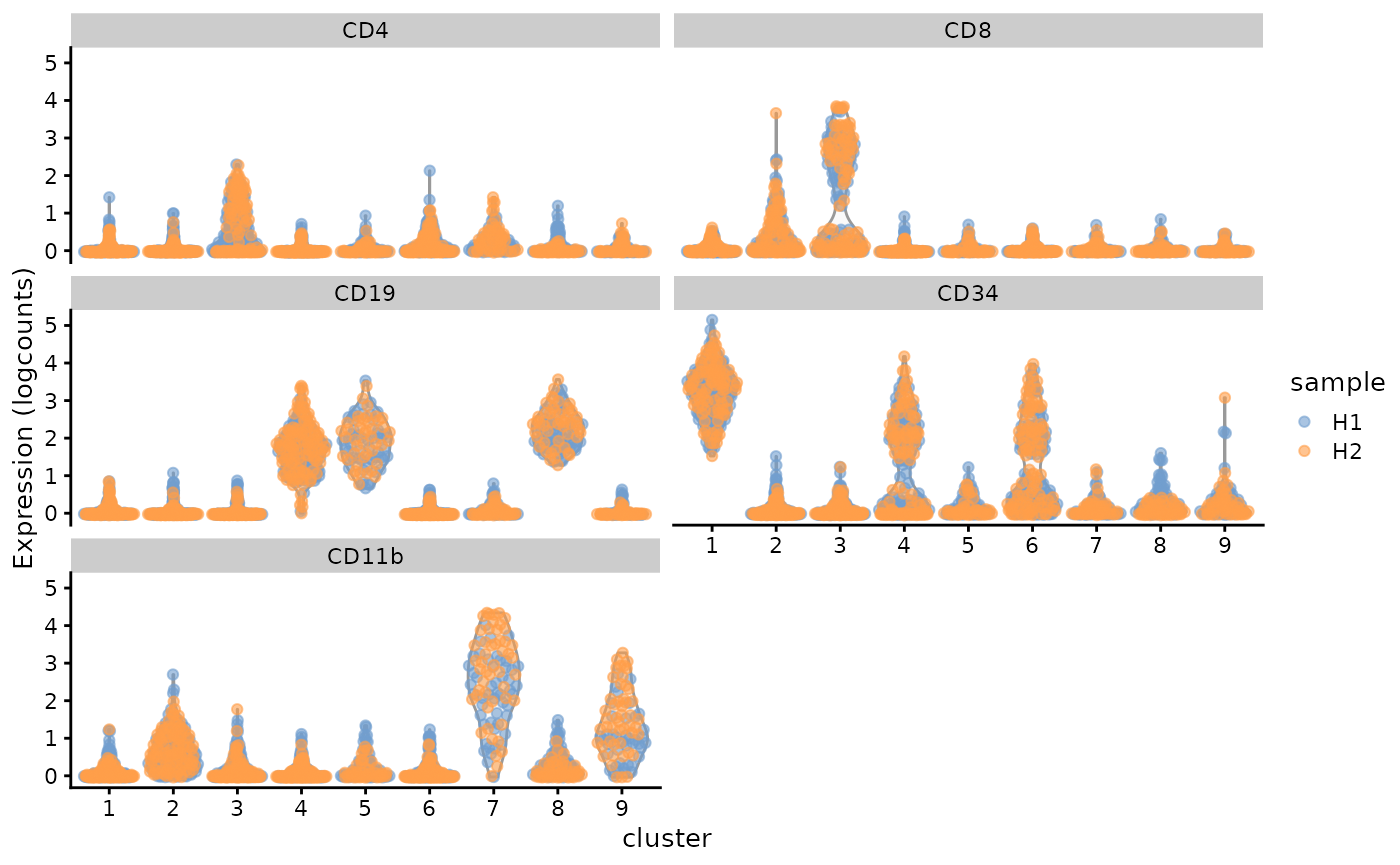
Or violin plot to see the distribution of their marker expressions. Note, the y-axis says “logcounts”, but the data is actually arcsinh transformed, not log transformed.
plotExpression(
sce, c("CD4", "CD8", "CD19", "CD34", "CD11b"),
x = "cluster", colour_by = "sample"
)
Session information
sessionInfo()
#> R version 4.5.1 (2025-06-13)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] data.table_1.17.8 bluster_1.18.0
#> [3] scater_1.36.0 ggplot2_4.0.0
#> [5] BiocSingular_1.24.0 scran_1.36.0
#> [7] scuttle_1.18.0 SingleCellExperiment_1.30.1
#> [9] SummarizedExperiment_1.38.1 Biobase_2.68.0
#> [11] GenomicRanges_1.60.0 GenomeInfoDb_1.44.3
#> [13] IRanges_2.42.0 S4Vectors_0.46.0
#> [15] BiocGenerics_0.54.1 generics_0.1.4
#> [17] MatrixGenerics_1.20.0 matrixStats_1.5.0
#> [19] SuperCellCyto_0.99.3 BiocStyle_2.36.0
#>
#> loaded via a namespace (and not attached):
#> [1] gridExtra_2.3 rlang_1.1.6 magrittr_2.0.4
#> [4] compiler_4.5.1 systemfonts_1.3.1 vctrs_0.6.5
#> [7] pkgconfig_2.0.3 crayon_1.5.3 fastmap_1.2.0
#> [10] XVector_0.48.0 labeling_0.4.3 SuperCell_1.0.1
#> [13] rmarkdown_2.30 UCSC.utils_1.4.0 ggbeeswarm_0.7.2
#> [16] ragg_1.5.0 xfun_0.53 cachem_1.1.0
#> [19] beachmat_2.24.0 jsonlite_2.0.0 DelayedArray_0.34.1
#> [22] BiocParallel_1.42.2 irlba_2.3.5.1 parallel_4.5.1
#> [25] cluster_2.1.8.1 R6_2.6.1 bslib_0.9.0
#> [28] RColorBrewer_1.1-3 limma_3.64.3 jquerylib_0.1.4
#> [31] Rcpp_1.1.0 bookdown_0.45 knitr_1.50
#> [34] FNN_1.1.4.1 Matrix_1.7-3 igraph_2.1.4
#> [37] tidyselect_1.2.1 abind_1.4-8 yaml_2.3.10
#> [40] viridis_0.6.5 codetools_0.2-20 lattice_0.22-7
#> [43] tibble_3.3.0 plyr_1.8.9 withr_3.0.2
#> [46] S7_0.2.0 evaluate_1.0.5 desc_1.4.3
#> [49] pillar_1.11.1 BiocManager_1.30.26 scales_1.4.0
#> [52] glue_1.8.0 metapod_1.16.0 tools_4.5.1
#> [55] BiocNeighbors_2.2.0 ScaledMatrix_1.16.0 locfit_1.5-9.12
#> [58] RANN_2.6.2 fs_1.6.6 cowplot_1.2.0
#> [61] grid_4.5.1 edgeR_4.6.3 GenomeInfoDbData_1.2.14
#> [64] beeswarm_0.4.0 vipor_0.4.7 cli_3.6.5
#> [67] rsvd_1.0.5 textshaping_1.0.4 S4Arrays_1.8.1
#> [70] viridisLite_0.4.2 dplyr_1.1.4 uwot_0.2.3
#> [73] gtable_0.3.6 sass_0.4.10 digest_0.6.37
#> [76] SparseArray_1.8.1 ggrepel_0.9.6 dqrng_0.4.1
#> [79] htmlwidgets_1.6.4 farver_2.1.2 htmltools_0.5.8.1
#> [82] pkgdown_2.1.3 lifecycle_1.0.4 httr_1.4.7
#> [85] statmod_1.5.1